治験は治療ではなく、試験。患者の正しい理解が治療薬開発を進める:ハイブリッドセミナー「治験について正しく知ろう」レポート

2025年7月27日(日)、セミナー「治験について正しく知ろう」をハイブリッド開催し、約350人(会場50名、オンライン300名)が参加しました。
治療薬の開発には、最後に治験があります。今回のセミナーは「治験」について正しい知識を得ることを目的とし、各方面の専門の先生方からご講演いただきました。
今回は、各先生の許諾を得て講演内容をビデオで公開いたします。
録画はDM-Familyチャンネルをご覧ください。DM-Family JAPAN - YouTube
(旧DM-Familyチャンネルから移転しました)
はじめに:日本の患者が「治験」を正しく理解しないと、治験をしてもらえないかもしれない

みなさん、今一度認識してください。治療薬開発には技術はもちろん、お金も、人も必要ですが、患者が治験に参加しないと薬はできません。そのためには治験を正しく知ることが大切です。
筋強直性ジストロフィーの治療薬は、現在、海外では10件の治験が実施されていますが、日本では1件であり、いわゆる「ドラッグ・ロス」の状況です。
「日本の患者が治験のことをよく知らない」という事実もあり、日本における国際共同治験が進んでいません。
しかし、海外では専門病院などで患者に治験のことを教えており、「治験は研究者にも患者にも試練のときである」と認識されています。
日本の患者も学び、賢くならねばなりません。
治験のキホン

臨床研究支援部 部長 中村治雅先生
DM-Familyからのお勧めポイント
・日本のお薬について取り扱う法律の中でも、「国民が、薬の有効性や安全性について理解を深める」ことがうたわれている
・臨床研究や治験のことを、患者にもよく理解してもらい、一緒に進めていくことが世界中で求められている
・治験は、世の中に出る前に「効き目があるか」「安全か」を人の体で確認する最終チェック
・治験が行われて世の中に出たからといって、その治療法が絶対に「安全」とは言い切れない。販売されて使えるようになった後も含めて、薬はみんなで育てていくもの
○患者の理解と協力が必要
薬機法*の第一条には、「国民は(中略)有効性及び安全性に関する知識と理解を深めるよう努めなくてはならない」とあります。
そして今、臨床研究や治験の方向性を考える国の会議で、重要なポイントのひとつとして「患者が理解し、協力してもらうことが大切である」とされています。
患者や市民の参画は社会全体からもとめられています。
*医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律
○薬ができるまで
薬は薬機法に基づいて規制されており、有効性や安全性を確認し、承認を受けなければなりません。薬を作るとき、基礎研究から応用研究(患者に使えるようになるまで、治験)がとても大変です。
基礎研究では、薬になりそうなものを探し出します。次に非臨床試験です。動物で有効性や安全性を調べます。これらでうまくいったものについて、ようやく人で試すことができます(いわゆる治験)。
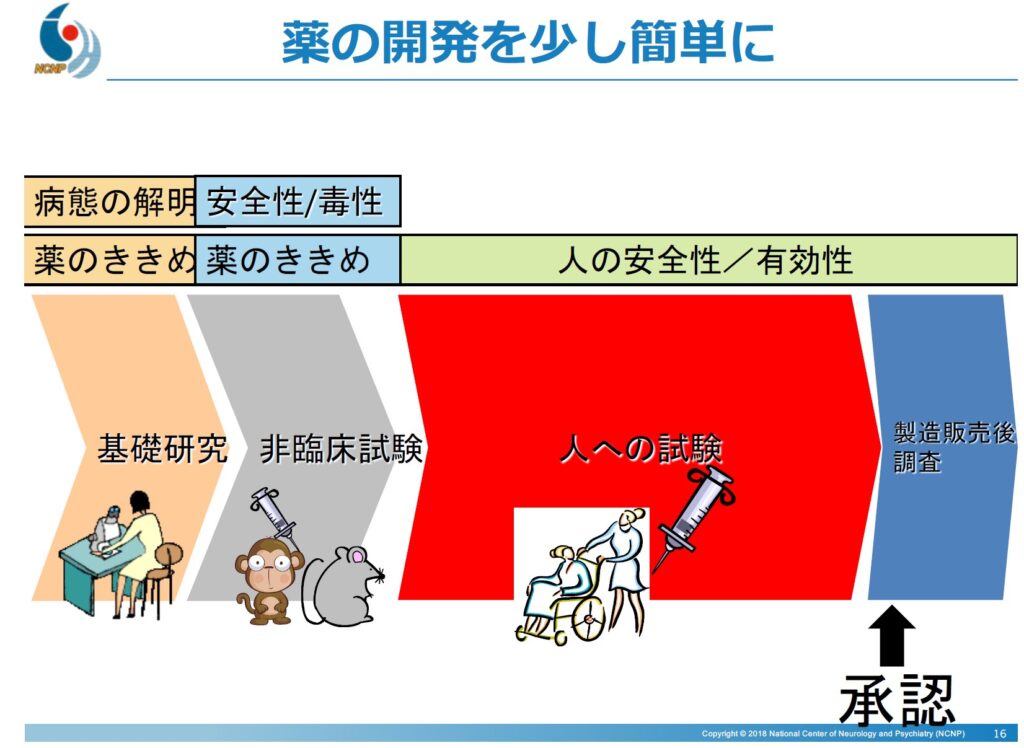
治験とは、国に医薬品の承認申請をするための試験です。
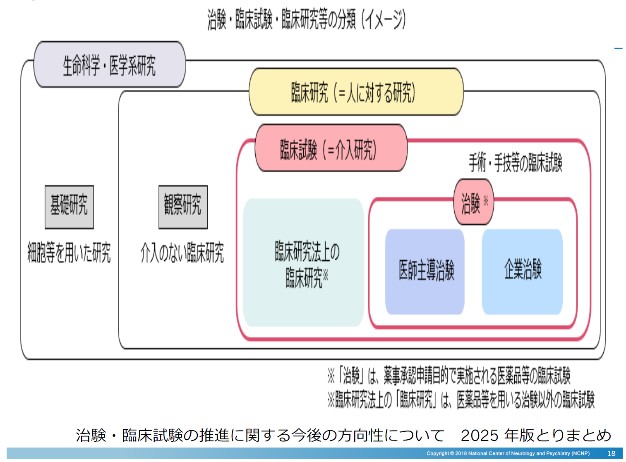
いろいろな言葉を整理すると以下のようになります。
- 臨床研究:人を対象にした様々な研究
- 臨床試験:普段の診療ではなく、何らかを実験的に患者に行う(介入する)研究
- 治験:臨床試験のなかで、医薬品の承認のために必要なデータを集める試験
なお、「治験」について誤ったイメージをお持ちの方もおられるかもしれませんが、決して怪しいものではありません。治験には倫理性や科学性、信頼性を確保して、治験に参加する人の人権や安全が守られる規則があり、GCP(Good Clinical Practice)といいます。参加する人の安全性が最優先されており、厳しい倫理的配慮がなされています。

*GCP (Good Clinical Practice)とは:「医薬品の臨床試験の実施の基準に関する省令」。 治験が適正かつ参加者の人権を守りつつ実施されるように、国が薬機法にもとづいて定めた規則。
治験の流れを簡易的にまとめると次のようになります。


第1相試験は初めて人に投与されるため、とても慎重に実施されます。また、長く使う薬の安全性をみるために、多くの患者で実施する長期投与試験(1年以上)も行います。
一般的にはこのような流れになりますが、筋肉の病気のような希少な疾患の場合は異なるところもあります。
ただ、基礎研究、前臨床試験、臨床試験などを経て使われるようになるまでに、一般的なお薬には10~17年ほど要し、基礎研究で検討した60万以上の物質のうち、承認され薬価が設定されるのは20ほどであるなど、薬を作ることは非常に大変であることも事実です。
○なぜ治験が必要なのか?
過去に薬害(サリドマイドなど)や深刻な副作用(キノホルムなど)が出たことがあります。本当にみんなが使っても大丈夫なのか、ということを調べないといけません。
また、動物実験で良い効果があっても、必ずしも人で同じ結果が出るとは限りません。やはり動物と人とは全く同じではありません。人での有効性と安全性が重要であるため、新しい薬が世に出る前の“最終チェック“として治験を行う必要があります。
ただし、治験の中に参加されている患者はとても限定的な人(他には何か合併症のない人や若い人など)、いわば限定的な検討であることから、治験だけでは絶対大丈夫とは言えず、不十分である可能性があることも理解する必要があります。
薬が世に出てからも患者や企業、研究者が情報を共有して、有効性や安全性に関するデータを蓄積し、良い薬に育てていく必要があります。
治験で薬の効果を測ることとは

DM-Familyからのお勧めポイント
・治験は、安全のために「試験を終えられる状態の患者」が対象
・治験には参加できない患者でも、承認・販売されれば、ほとんどの患者が治療薬を使える
・治験は数人の思い込みではなく、本当に効果があるかどうかを見極める「比較」を行う
・本当に効果があったかどうかを確認する「評価指標」には、自然歴研究や患者登録のデータが必要
○薬が「効く」とは
熱や痛みの軽減だけではなく、病気の進行が遅くなることも薬の効果です。また、頭痛薬により膝の痛みが緩和するなど、主な効果のほかの二次的な効果が現れることもあるため評価が必要です。
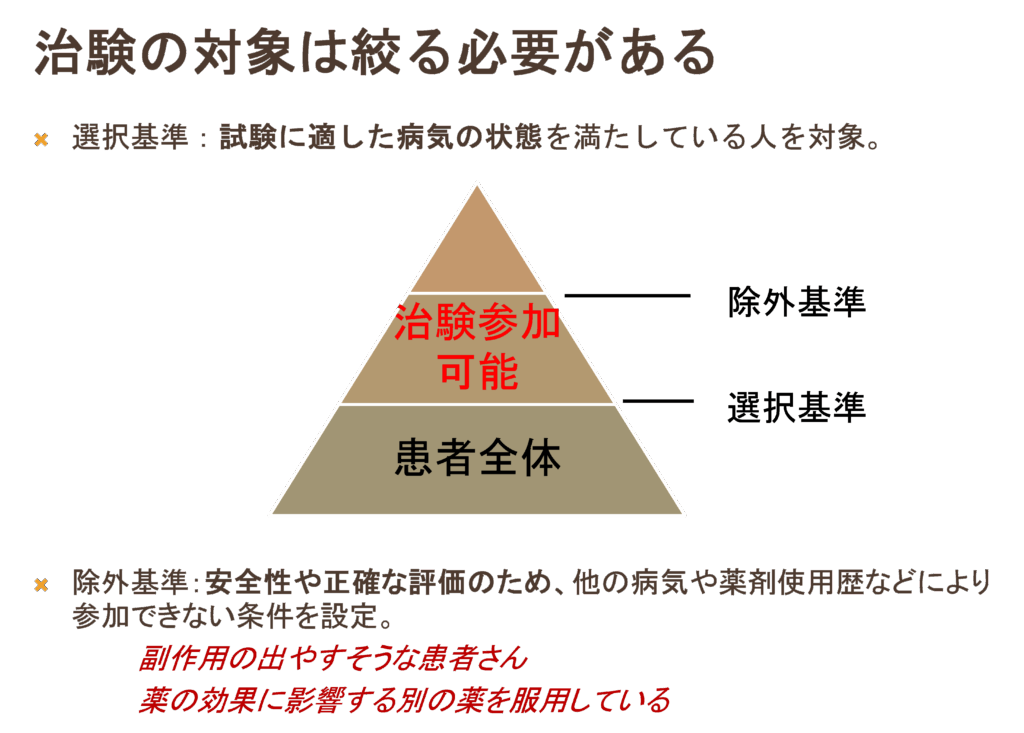
○治験に参加出来る人はどんな人か
本当に効いたのかを確認することは難しいことであり、そのために「治験」が必要です。各段階で目的と方法が異なり、段階的にリスクを減らして実施します。
治験はどんな人でも対象になるわけではなく、短期間で効果が期待できる人や、他の影響をできるだけ避けられそうな人で検討されます。試験に適した病気の状態を満たしている人を対象とし(選択基準)、安全性等を考慮して参加できない条件(除外基準)が設定されています。
○どのように治験を行うのか
基本的には「比較」を行います。具体的には、薬を飲む人と飲まない人の2つにランダム(無秩序)で分けて比較します。また、思い込みを排除するため、患者も医療者も、どの薬を割り当てられているのかわからない状態で試験を行います(「二重盲検試験」といいます)。
また、『効いた』といえるためのルールが必要です。結果について統計学的な有意差を確認して効果を確認します。そのためにも母集団をある程度コントロールすることが大切です。

○どうやって効果を評価するのか~評価項目~
治験では「主要評価項目」と「副次評価項目」というものが設定されます。それぞれ、症状の改善度など最も重要な効果を図る指標、生活の質など補助的な効果を図る指標です。治験は目的に応じて「事前に」評価項目を決めます。
例えば、糖尿病の治療薬の治験では、HbA1Cという評価項目があります。これはHbA1Cと糖尿病の合併症のリスクの関係が判明しているために用いられます。しかし、筋強直性ジストロフィーではそのような評価指標がない状況です。この病気の治験は少しずつ始まっていますが、評価項目はさまざまで手探りの状態です。
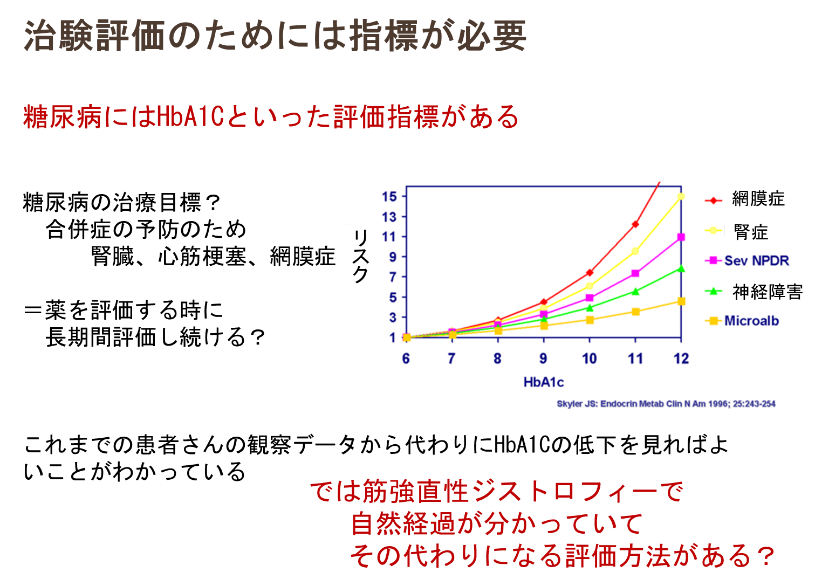
そのためにも薬を使っていない患者のデータが大切であり、自然歴研究*が進められています。自然歴研究は全世界で実施されています。同じ指標でデータを蓄積することは、国際共同治験実施の基盤になるものであり、すでに実際の治験で活用されています。
*治験を想定した患者を対象とし、患者の状態などがどのように変わっていくのかを調べる研究

なお、参加基準がない患者登録「Remudy(レムディー)」も参考になり、適格性基準や評価項目の事前検討に役立ちます。
また、患者のQOL(生活の質)評価も良く利用されます。アンケートでデータ収集しますが、もとのアンケートが英語であるため、定められた方法で日本語訳されたものを使用しています。(英語版と異なる解釈にならないように、日本語訳作業は大変重要です)
○筋強直性ジストロフィーの治験の実際は
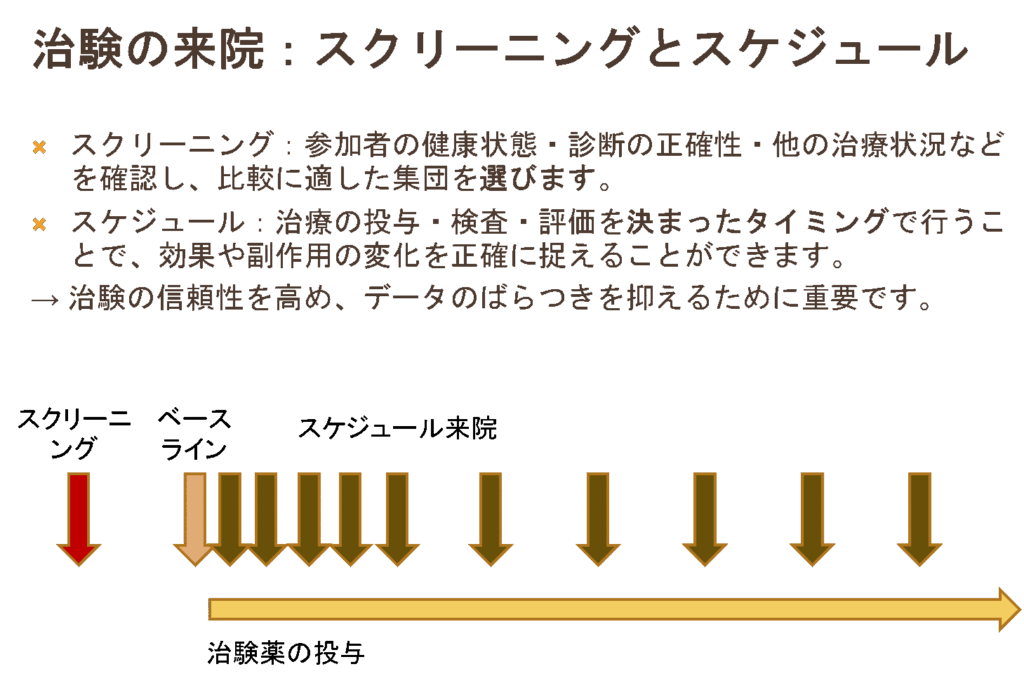
参加する場合、治験被験者は説明を聞いて同意したあと、診断の正確さや状態(さきほどの選択基準に合致しているかなど)を評価(スクリーニング)されます。
その後、薬の投与前の評価(ベースライン)を受け、投与後に定期的に通院してさまざまな評価を受けます。
治験の信頼性を高めるために通院スケジュールはしっかりと管理されています。
最近の治験(第2相、https://jrct.mhlw.go.jp/latest-detail/jRCT2051190069)での具体例を紹介します。あくまでも一例であり、試験によって異なります。
■適格基準(主たる選択基準)
![]() 同意取得時に20歳以上55歳以下の患者。性別は問わない。
同意取得時に20歳以上55歳以下の患者。性別は問わない。
![]() DMPK遺伝子変異が確認されており、CTGリピート長が100以上と診断されている筋強直性ジストロフィー患者。
DMPK遺伝子変異が確認されており、CTGリピート長が100以上と診断されている筋強直性ジストロフィー患者。
![]() スクリーニング時に6分間歩行試験で25メートル以上歩行できる患者。
スクリーニング時に6分間歩行試験で25メートル以上歩行できる患者。
![]() 本治験の参加にあたり十分な説明を受け、理解し、本人による文書同意が得られた患者。
本治験の参加にあたり十分な説明を受け、理解し、本人による文書同意が得られた患者。
■主たる除外基準(抜粋)
![]() スクリーニング検査で心電図上心臓伝導障害をもつ患者および心臓ペースメーカーを装着している患者。
スクリーニング検査で心電図上心臓伝導障害をもつ患者および心臓ペースメーカーを装着している患者。
![]() スクリーニング検査で認知機能低下のある患者。
スクリーニング検査で認知機能低下のある患者。
![]() スクリーニング検査において、肝機能(AST、ALT、γ-GTP)検査値が年齢、性別における基準値上限の3倍を超えた患者。あるいはスクリーニング時に血清クレアチニン値が基準値の2倍を超えた患者。
スクリーニング検査において、肝機能(AST、ALT、γ-GTP)検査値が年齢、性別における基準値上限の3倍を超えた患者。あるいはスクリーニング時に血清クレアチニン値が基準値の2倍を超えた患者。
![]() スクリーニング前30日以内にメキシレチン、フェニトイン、カルバマゼピン、プロカインアミドなど筋強直に影響を与える薬剤を使用した患者、あるいは治験のすべての評価が終了する前に当該薬使用が予想される患者。
スクリーニング前30日以内にメキシレチン、フェニトイン、カルバマゼピン、プロカインアミドなど筋強直に影響を与える薬剤を使用した患者、あるいは治験のすべての評価が終了する前に当該薬使用が予想される患者。
■主たる評価項目
![]() 安全性評価項目
安全性評価項目
(すべての有害事象の頻度集計及び発現割合)とする。
→主たる項目は通常1つです。この治験は第2相であるため、まずは安全性を重視します。
■副次的な評価項目
![]() 骨格筋でのスプライシング異常の改善度
骨格筋でのスプライシング異常の改善度
![]() 針筋電図検査におけるミオトニアスコアの推移
針筋電図検査におけるミオトニアスコアの推移
![]() 筋力計による筋力評価
筋力計による筋力評価
![]() 6分間歩行における歩行距離による下肢機能
6分間歩行における歩行距離による下肢機能
![]() 神経筋疾患患者のQOLアンケートの評価
神経筋疾患患者のQOLアンケートの評価
![]() 臨床全般印象度(CGI)の評価
臨床全般印象度(CGI)の評価
![]() クレアチンキナーゼ(CK)の減少量
クレアチンキナーゼ(CK)の減少量
第2相試験であるため、筋生検や筋電図があります。このように日頃の診療とは異なることも実施します。
○薬が安全であることを確かめるために
予期できない副作用を見つけるために、治験期間中の有害事象(望ましくない出来事)を全て報告する必要があります。
例えば、風邪や骨折、他の病気での受診など、これら全ての情報を記録して、薬のリスクを評価する必要があります。
治験と倫理~患者の役割を大切にするために

DM-Familyからのお勧めポイント
・医療の発展のためには、人の体を使う治験が必要
・治験に参加した患者の権利を守るために、さまざまな規制がある
・第二次世界大戦中の人体実験のようなことを二度としないために、「ヘルシンキ宣言」がある
○よりよい治験を行うために患者が果たす役割がある
さまざまな場所で患者が治験の仕組みづくりに参画するようになっています。
治験への参加だけではなく、「どうやって薬を評価するのか」について、患者が研究者や開発者とともに考えていくことが世界的な認識となっており、患者自らが声をあげることで、自分たちが考えていることを反映できるようになっています。
○治験において患者を守るしくみ
医療と医学の発展には人を対象とする研究が欠かせません。黙って座っているだけで新しい治療法が来ることはないわけです。
医療従事者とともに患者もがんばらないと治療法は得られないです。
治験では被験者の権利を守り、安全性を高め、その利益を公正に共有するため、倫理原則やGCPなどの規制があります。
治療法開発時にかかる費用を節約し、無駄を省くことで「治験のエコシステム*」を構築するためには、研究者や企業の視点ではわかりにくいので、患者の視点が必要になっています。
*国民にいち早く治療薬を届けるため、製薬企業、医療機関、規制当局、患者など、あらゆる関係者が協力して効率的に治験を行うシステム
○人での実験は欠くことはできないなかで、倫理原則は発展し続けている
新しい薬の開発には人での実験を欠くことはできません。
第二次世界大戦中の人体実験などの酷い経験を繰り返さないよう、世界医師会において、人間を対象とする医学研究の倫理的原則として「ヘルシンキ宣言(1964年)」がなされています。
その後も、倫理的に問題がある研究が指摘されるごとに、この倫理原則が強化されています。
2024年のヘルシンキ宣言改訂においては、患者の研究参画が大きく取り上げられました。
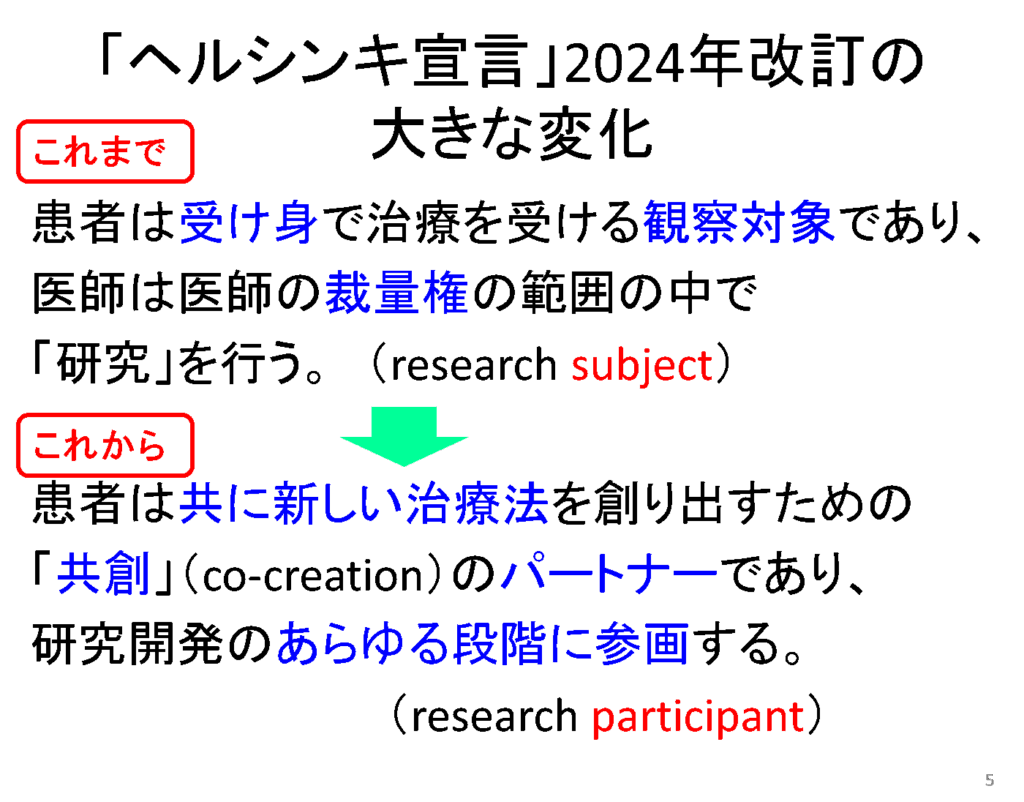
それまでは患者は受け身でしたが、これからは一緒に新しい治療法を開発するためのパートナーであり、研究のあらゆる段階に参画することとなり、大きく変化しました。
これにより、よい治験のデザインや、地域・世界で薬を必要とする人々に届けるにはどうしたらよいか、などについてみんなで考えていくようになっています。
また、ヘルシンキ宣言は、医師だけではなく、看護師や薬剤師、理学療法士、入退院の支援にあたる人々をはじめ、それらから成るチームや組織に支持されることが望ましい、と改訂されました。
○日本の規制の概要
日本には研究の規制は多くのものがありますが、以下の3つについて紹介します。
■GCP省令
![]() 薬機法に基づき、承認申請(製造販売)を目的とする治験等を適正に行うためのルール
薬機法に基づき、承認申請(製造販売)を目的とする治験等を適正に行うためのルール
■臨床研究法
![]() 薬機法の有効性・安全性を明らかにするが申請目的がない
薬機法の有効性・安全性を明らかにするが申請目的がない
![]() 必ずしも承認申請にターゲットを絞っていないが有効性等を見たいもの
必ずしも承認申請にターゲットを絞っていないが有効性等を見たいもの
![]() 細胞を加工して人に投与するような試験も含む
細胞を加工して人に投与するような試験も含む
![]() 厚労省の認定を受けた臨床研究審査委員会で検討されており、ある程度安全性は守られる
厚労省の認定を受けた臨床研究審査委員会で検討されており、ある程度安全性は守られる
![]() 将来的には承認申請のデータにも活用できる可能性がある
将来的には承認申請のデータにも活用できる可能性がある
■人を対象とする生命科学・医学系研究に関する倫理指針
![]() 上記の2つ以外
上記の2つ以外
![]() 人の情報やサンプルの分析や、手術等に関する研究など
人の情報やサンプルの分析や、手術等に関する研究など
![]() 前述の自然歴研究も該当
前述の自然歴研究も該当
いずれもヘルシンキ宣言を基にしています。
また、これら研究は非専門家(一般市民や法律の専門家等)も含む倫理審査委員会の目を通って進められていることも理解しておく必要があります。
○ヘルシンキ宣言 ~患者の権利とは~
前述のとおり、「人を対象とする医学研究の倫理原則」です。最も重要な原則は「研究対象者の権利と利益は、研究の目的より優先される」ということです。内容は、
![]() 倫理審査委員会の審査、インフォームドコンセントの内容。十分な理解の上での同意が必要
倫理審査委員会の審査、インフォームドコンセントの内容。十分な理解の上での同意が必要
![]() 良くない結果も発表、研究の情報は公開データベースに登録する
良くない結果も発表、研究の情報は公開データベースに登録する
![]() 研究計画書や論文には「ヘルシンキ宣言を守る」と記載
研究計画書や論文には「ヘルシンキ宣言を守る」と記載
![]() 情報は漏洩してはならず、プライバシーも守られる
情報は漏洩してはならず、プライバシーも守られる
![]() プラセボ対照試験*は、既存の有効な治療法がない場合にのみ実施されるべき(例外的に既存の有効な治療法がある場合にはプラセボ群に割り付けられた場合のリスクが厳しく制限される)
プラセボ対照試験*は、既存の有効な治療法がない場合にのみ実施されるべき(例外的に既存の有効な治療法がある場合にはプラセボ群に割り付けられた場合のリスクが厳しく制限される)
などです。
ヘルシンキ宣言は5枚ほどの量なので、手軽に読み、理解することができます。研究に参加する際には是非お読みいただくことが重要です。
https://www.med.or.jp/dl-med/wma/202410kaitei_helsinki_j.pdf
*プラセボ対照試験とは、試験薬と形状は同じだが薬効のないプラセボ(偽薬)を受けるグループと、試験薬を受けるグループに、患者をランダム割り振って、試験薬に本当に効果があるかどうかを調べる方法。患者は自分がどちらのグループに割り振られるかわからないということを説明されて理解した上で参加する。
患者には最も良い治療を受ける権利や、プライバシーや患者の安全を守られる権利などがあります。声を出し続ければ届きます。思いを言葉にし、仲間とともに活動を継続することが大切です。
医療基盤開発研究所(Ji4pe)の生命倫理ワーキンググループでは、患者市民主導でヘルシンキ宣言についての勉強会を重ねて、宣言の2024年改訂に寄せて、この宣言には含まれない、患者市民目線の倫理原則を「患者・市民の研究倫理宣言」として世界に発信されています。患者市民が自ら声をあげれば世界に届くという実例なので、是非ご確認ください。
http://cont.o.oo7.jp/52_3/p463-74.pdf
PMDA における新薬の審査について

DM-Familyからのお勧めポイント
・PMDAは薬の有効性や安全性等について科学的な判断をしている
・新薬の審査は、専門知識を有する審査員からなるチームで担当
・新薬の審査のスピードは世界でもトップクラス
・PMDAは、患者への情報提供・周知や患者からの情報収集を主軸とする「PMDA患者参画ガイダンス」の発出以降、患者参画の取り組みを推進している
○PMDAとは?
PMDAは医薬品医療機器総合機構(Pharmaceuticals and Medical Devices Agency)の略称で、厚生労働省(厚労省)所管の独立行政法人です。元々国で行っていた新薬の審査や安全対策等の業務を行うことを目的に設立されています。
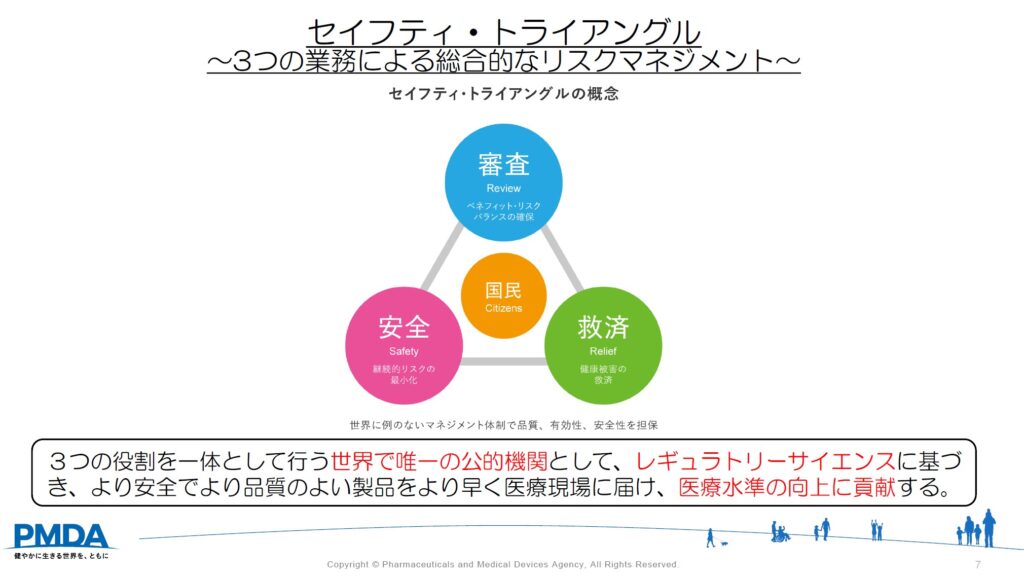
PMDA法と薬機法に基づき、主に次の3つの業務を行っています。
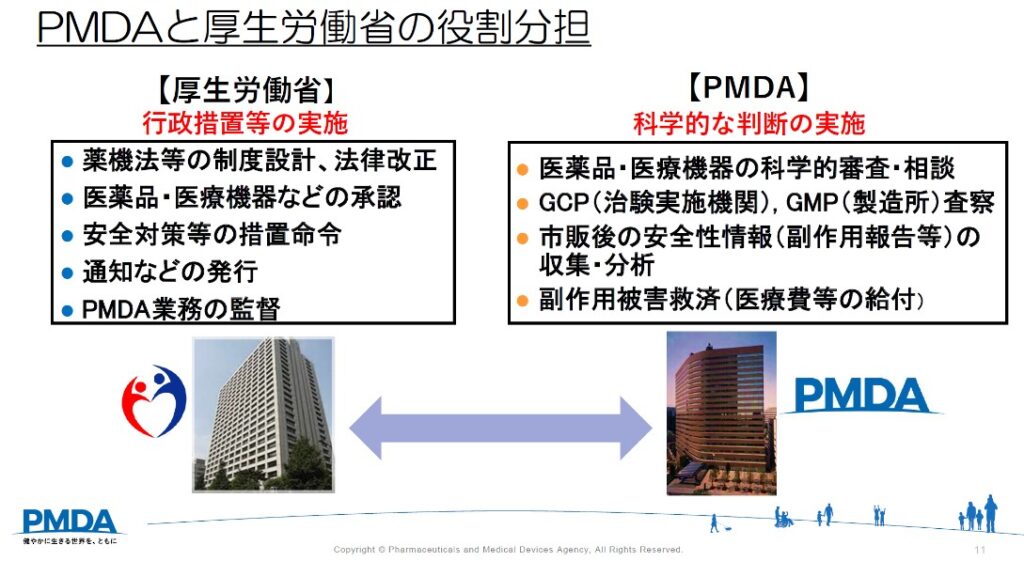
■健康被害救済
医薬品等による健康被害の迅速な救済
→医薬品等の副作用により、入院等が必要になった場合に、金銭的に救済する制度
■承認審査
医薬品等の品質・有効性・安全性について助言、承認審査
■安全対策
市販後の安全性情報の収集、分析、提供
→医薬品等の承認後に新たな副作用が報告された場合等に追加の安全対策措置を検討する
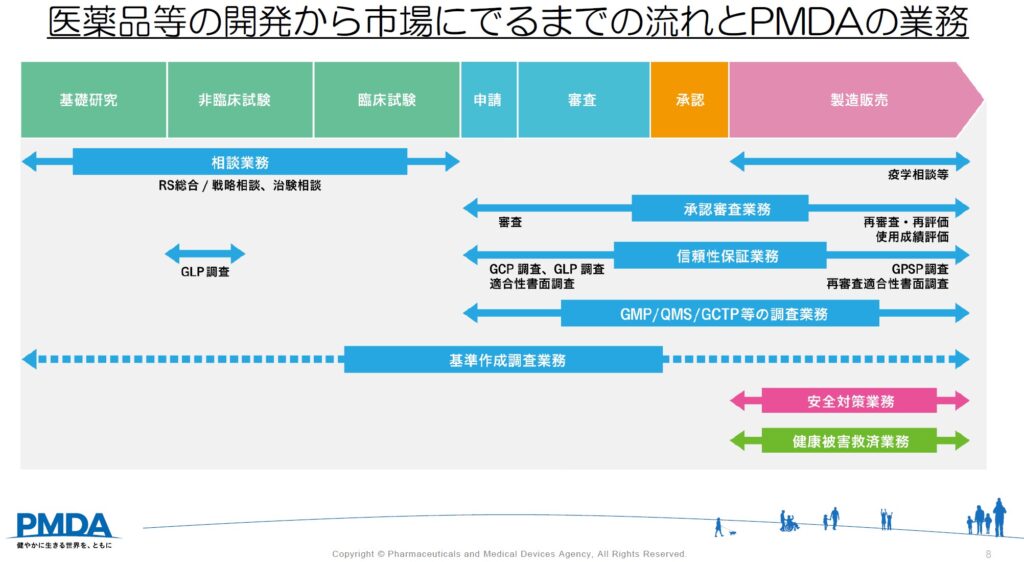
PMDAは医薬品の開発から販売後まで関わっています。例えば、相談業務では、企業等が実施を予定している臨床試験デザイン等について相談を受け、承認に向けてより効率的かつ適切に開発が進むようアドバイスを行います。新しい薬をできるだけ早く世に届けるため、このように開発早期の段階から関わっています。
このセミナーでは新薬の申請から承認までどのように審査を行っているのか概要を説明します。
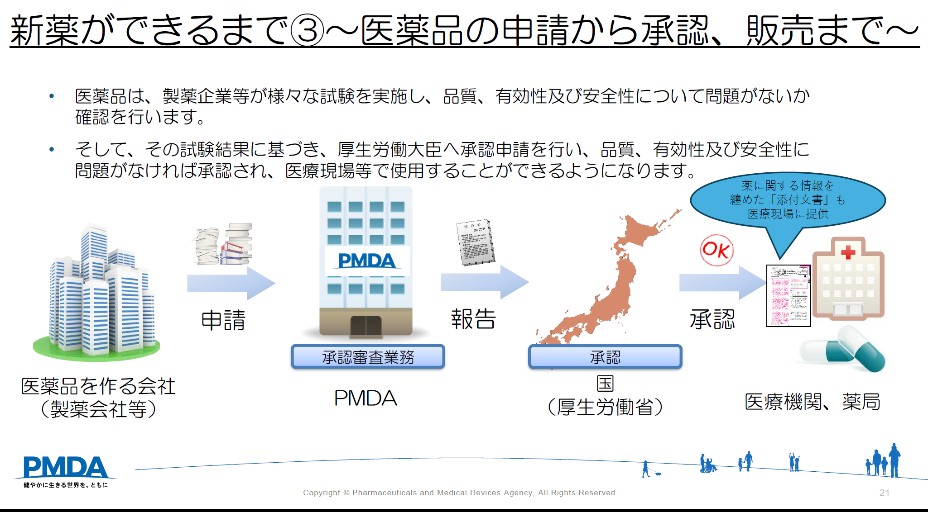
○新しい治療薬(新薬)の審査
医薬品は大きく分けると「医療用医薬品」と「一般用医薬品」があり、医療用医薬品の種類として「新医薬品(新薬)」、「後発医療用医薬品」、「バイオ後続品」があります。
PMDAの新薬審査部門(再生医療等製品を審査する部門を除く)は大きく6つの部に分かれており、筋強直性ジストロフィー等の神経難病にかかる新薬の審査は新薬審査第三部にて行っています。
新薬は基礎研究を経て、非臨床試験にて動物等で効果と安全性を評価し、臨床試験(治験)が行われます。
そこで得られた試験結果に基づき、製薬会社等から厚労大臣へ承認申請され、PMDAで審査します。
PMDAは、審査の結果を厚労省に報告し、有効性や安全性等に問題がなければ承認されます。
新薬が承認された後、保険の適用が認められると医療機関や薬局で使用されることになります。
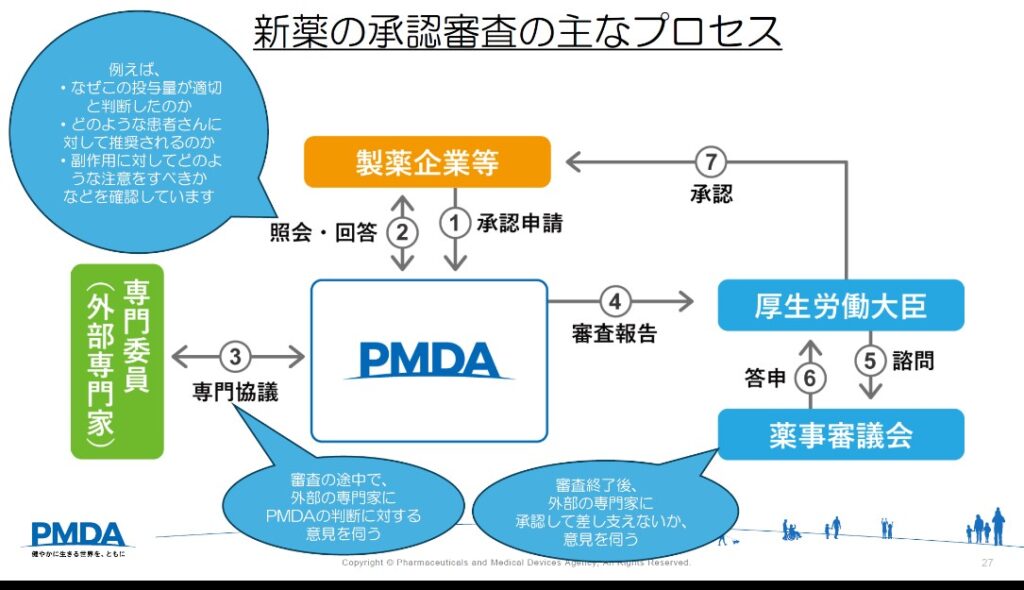
新薬の審査は、まず製薬会社等から承認申請(下図①)する際に提出された膨大な資料を読み解くところから始まります。
提出された資料の不明点等について申請企業に照会し回答を得ます。この照会・回答を繰り返して審査を進めます(②)。
承認の可否等について概ね判断できた段階で、外部の専門家(専門委員)に対して意見を伺います(③)。
専門委員の意見を踏まえて、審査報告書を取りまとめ、厚労省に対して審査結果を報告し(④)、厚労省にて、薬事審議会に諮問し、外部の専門家に意見を改めて伺い(⑤)、その結果(答申)を受けて(⑥)、厚労大臣によって承認されます(⑦)。
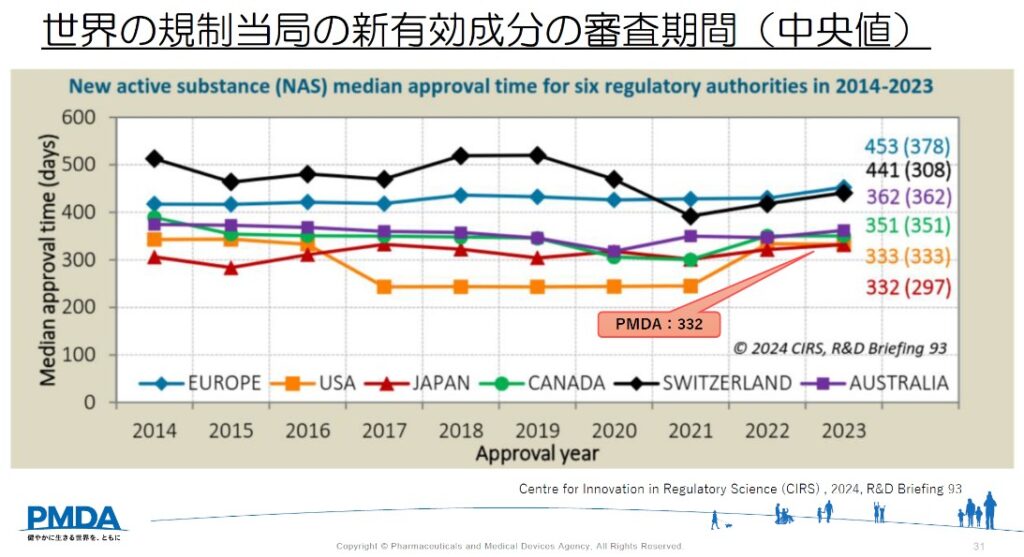
新薬の審査期間は基本的に12か月を目標として進められますが、患者数が少なく、重篤で、他に治療法がない疾病に対する医薬品(希少疾病用医薬品)では、9か月を目標に審査が進められます。
審査期間が長いと思う方も多いかもしれませんが、世界の中でも日本の審査のスピードはトップクラスです。
ドラッグ・ロス、ドラッグ・ラグの問題は、審査にかかる期間ではなく、日本で開発されない、日本での開発に時間を要していることが主な要因と言えます。
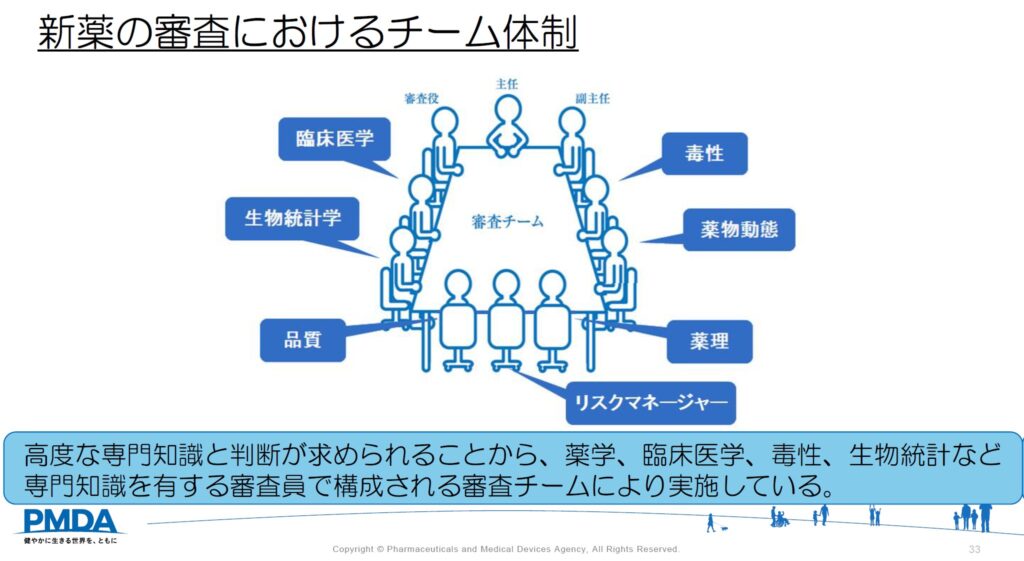
承認申請時に提出される資料には、薬の品質に関する資料や、非臨床試験データ、臨床試験データに関する資料等が含まれます。高度な専門知識と判断が求められるため、様々な専門知識を有する審査員で構成されるチームで審査を行っています。
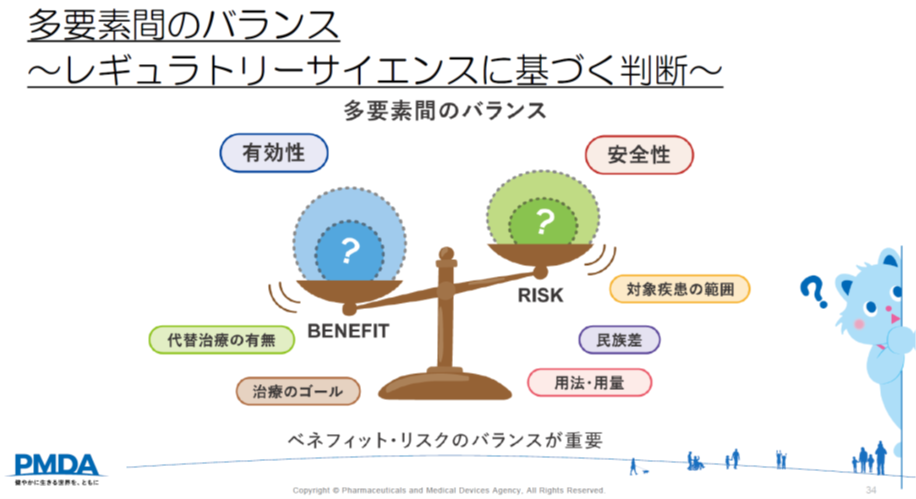
審査にあたっては、「何点以上が合格」といった定量的に評価できるものではなく、さまざまなデータを確認し、新薬の有効性、安全性を多角的に評価し、ベネフィット・リスクバランスを判断します。
審査の着目点は、薬の身近な疑問と似ており、「副作用やその頻度は?」「保存方法は?」「他の薬との併用は大丈夫?」などの疑問に対して、データに基づいて確認しており、申請企業の主張に対して、客観的に判断することがPMDAの重要な役割です。
なお、審査結果は審査報告書としてまとめられ、PMDAのウェブサイトで公開されています。このウェブサイトでは、薬の基本情報がまとめられた添付文書や患者向医薬品ガイドも公開されています。
希少疾病用医薬品は、患者が少ないこと等から、他の薬と比較して有効性・安全性を十分に評価するための臨床試験を行うことが難しく、審査も難しいです。その中でPMDAの審査員も、より良い医薬品がより早く使えるように、日々悩み、検討しています。
○患者参画の取り組み
PMDAは、患者と相互でコミュニケーションを図るためのPMDAの取り組みを検討すること等を目的に患者参画ワーキンググループ(WG)を発足させ、意見交換しています。
患者への情報提供・周知を行うために、このような講演等を通じて薬事制度の理解・普及を促進する取り組みを行っています。
また、最近では、患者がどのようなことに日々困っているのか、患者の声を直接聴き、業務に反映することが大切と考え、患者との意見交換会等の実施の検討も行っています。
質疑応答
本セミナーの講演後、参加者からの質問を受け、演者の先生方から回答いただきました。その一部をご紹介致します。
-
わたしの家にいる患者は、治験に参加できる状態ではない。絶望し、がっかりした。
-
治験に参加できなくても、治療薬が承認されれば、投薬を受けられる可能性があります。治験は「試験」で「治療」ではないことを理解してください。
-
筋強直性ジストロフィーの評価指標について、研究は進んでいるのか?
-
いろいろと研究はされている。企業では独自に工夫した指標を試しながら使っている。検査も進んでいるが、治験で使うにはもう少し時間がかかる。
-
外国の製薬企業が日本で治験を実施するということは、承認される一歩手前という理解でよいか?
-
臨床試験の内容、位置づけによる。試験の目的について説明を受けていると思うが、医薬品の有効性や安全性を検証することを目的とした臨床試験(第3相試験)であれば結果次第ではあるが、ステージとしては承認申請される段階になると思う。
-
臨床試験の場所はどこで確認したらよいか?治験の場所が遠い場合はどうしたらよいか?
-
国内で臨床試験が始まっていればjRCT(臨床研究等提出・公開システム、https://jrct.mhlw.go.jp/)、海外のみで臨床試験が行われているのであればClinicalTrials.gov(https://clinicaltrials.gov/)で確認できます。
筋強直性ジストロフィーについてはDM-Familyのサイトを参考に、ClinicalTrials.govで検索するとよいでしょう。
ただ、近くの場所ですぐに実施出来るようにはならないと思います。特に希少疾患の場合は国内で数か所ということも多い。なぜなら、治験を実施する病院は、実施する企業が選定するからです。
なお、現段階においては、筋肉の病気を評価するには理学療法士の評価が必要であるため病院にいかざるを得ません。
-
治験で除外基準にあたる人は、承認された薬は使えないのか?
-
普通は使用できると思う。
ただ、年齢によって(小児などで、適当な投与量の治験データがその時点ではない場合など)使えない場合があるかもしれません。
最後に
本セミナーの開催にあたり、米国のAvidity Biosciences Inc.様より資金をご提供いただきました。この場をお借りして御礼申し上げます。誠にありがとうございました。
Avidity Biosciences Inc.では、現在、筋強直性ジストロフィー治療薬「デルデシラン」の開発が進められており、国際共同治験第3相「HARBORTM試験」を実施しています。日本語での詳細は当会サイトをご覧ください。
また、本セミナー終了後に、Avidity Biosciences Inc.から筋強直性ジストロフィーの患者と家族に向けたメッセージが送られ、「HARBORTM試験の結果に基づき、2026年後半から米国、EU、日本を含む地域でデルデシランの販売承認申請を開始する予定です。」と伝えられました。
メッセージ全文はこちらをご覧ください。

