医薬品は実際にどのように承認審査が進められるのか:ウェビナー「PMDAを知ってみよう」レポート

PMDA(独立行政法人 医薬品医療機器総合機構)を知っていますか?
2024年11月17日(日)、ウェビナー「PMDAを知ってみよう」をオンラインで開催し、約70人が参加しました。
PMDAは「独立行政法人 医薬品医療機器総合機構」の略称で、医薬品や医療機器の審査などを行っている機関です。医薬品が承認されたニュースでも厚労省の名前は登場するものの、PMDAの名前はなかなか表に出てこないので、ご存じない方もいらっしゃるのではないでしょうか。
今回は、PMDAで働く医師の手塚瞬先生から、医薬品が医療現場に届くまではどのような過程を経ているのか、その中でPMDAがどのような業務を行っているのか、どのような視点で審査をしているのかを中心にご講演いただきました。
【お断り】今回のご講演内容やスライドにつきまして、手塚先生ご本人の見解や検討中の項目を含んでおり、必ずしもPMDAの公的見解ではないことにご留意ください。

主任専門員(臨床医学担当) 手塚瞬先生
1.はじめに
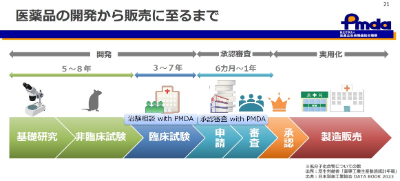
医薬品の開発から販売に至るまで
新しい医薬品の開発から販売に至るまでには長い時間を要します。開発は、基礎研究→非臨床試験→臨床試験と進んでいきますが、一般的には10~15年が必要となります。 臨床試験で開発医薬品の有効性及び安全性が示された後に、製薬企業が承認申請を行い、PMDAで審査を行い(審査期間は6カ月~1年)、承認して差し支えないと判断された場合、厚生労働省が承認し、販売され、医療現場で使えるようになります。
薬の候補物質のうち、臨床試験に到達するものは10,000分の1程度であり、承認されるものは25,000分の1程度です。薬の候補物質が見つかってニュースに取り上げられることはあるものの、その後、有効性や安全性が期待できる結果が得られず、開発が断念されたようなニュースはなかなか取り上げられません。実際には、臨床試験をしてみたら残念ながら効果が認められなかった、ということは非常に多いのです。したがって、臨床試験などで薬の候補の有効性や安全性を調べないと、効き目がないもの(時には、使わなかったときより病気が悪くなるもの)を間違って使ってしまうことになります。
薬の効果や副作用をヒトで調べる
臨床試験では、ヒトに対する治療効果や副作用を確認します。
なお、臨床試験は「ヒトに対する試験一般」のことを指し、治験は「候補薬を用い、国の承認を得るためデータを集める臨床試験」のことを指します。臨床試験では、主に、有効性、安全性、体内での薬の動きを調べます。
臨床試験には3つの段階(相)がある
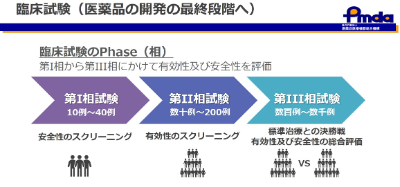
まず、第Ⅰ相試験では、少数のヒトに投与し、安全性を評価します。第Ⅱ相試験では有効性が期待できるかどうかを評価し、第Ⅲ相試験では現在行われている標準治療との比較を行うことが一般的です。
治験にはルールがある
治験は「医薬品医療機器等法」という法律に基づいて実施されますが、この法律に基づいて定めたルールも守る必要があります。このルールは「GCP*」と呼ばれ、国際的に決められています。

このルールに沿った臨床試験が製薬企業等で行われ、品質、有効性及び安全性に問題ないことが確認されると、国(厚生労働省)に承認申請され、PMDAで審査し、PMDAでの審査結果に基づき厚生労働省で承認され、医療現場で使用できるようになります。
*GCP:Good Clinical Practiceの略
2.PMDAとは?
PMDAは2004年に設立された、厚生労働省管轄の独立行政法人です。科学的判断に基づき、医薬等の評価を行っています(厚生労働省は、この判断結果を基に行政を担っています)。
より安全でより品質のよい製品をより早く医療現場に届け、医療水準の向上に貢献することを目的としています。

「薬」に求められることは人によって様々であり、安全性、品質、有効性、利便性、価格等が挙げられますが、PMDAが評価していることは主に安全性、品質、有効性です。
なお、PMDAで働いている職員は約1,500名で、薬学の背景を有する職員が多いですが、背景は様々で、臨床担当と呼ばれる医師については、約50名が勤務しています。
国際協調を踏まえ、評価方法も進歩している
PMDAをはじめ、アメリカのFDA*1、ヨーロッパのEMA*2、各国の製薬企業団体等が参画する医薬品規制調和国際会議(ICH)という組織があり、さまざまな評価やガイドラインを作っています。日本におけるGCPもICHのガイドラインに基づくものです。
*1 Food and Drug Administration:アメリカ食品医薬品局
*2 European Medicines Agency:欧州医薬品庁
3.医薬品承認審査の実際
医薬品の審査は機械的な書類仕事ではない。
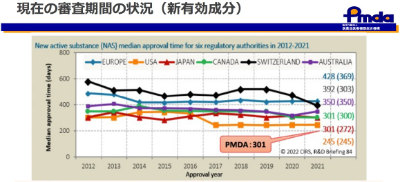
実は、審査スピードは世界最速レベル!
「審査は事務作業。書類が揃っていれば1カ月ほどで終わるのでは?」というような声をいただくこともあります。
医薬品の審査では、機械的な審査業務ではなく、臨床試験の成績や疾患の病態、医療現場の状況を踏まえ、中身の審査を行います。企業へのデータ照会や専門委員への意見伺いなどもあり、PMDAとしての結論を付けるためには時間が必要です。なお、ここ10年間では審査スピードは世界でも最速レベルになっています。
PMDAは薬の開発段階から関わっている
PMDAは承認審査だけではなく、開発においては企業やアカデミア(大学病院など)の治験の相談、販売後においては安全対策を行っており、薬のいわゆるライフサイクル全体に関わっています。特に、医薬品の審査にあたっては、評価可能な臨床試験成績があることが重要で、そのためには、臨床試験の計画時点からPMDAと企業とで相談を行うことがほぼ不可欠であり、審査と同様に相談業務の意義は大きいです。
どのように審査業務は進められるのか?
承認申請後、面談や必要事項の照会を行い、審査報告書を作成していきます。
審査報告書の第一版が確定した後に、PMDAが委嘱している外部専門委員とPMDAの審査方針について専門協議を行い、その結果を踏まえ、審査報告書が完全に確定します。
その後、厚生労働省が事務局を行う部会で審議され、承認へ進んでいきます。ここまでで通常の審査制度の場合は概ね1年かかります。
なお、ドラッグ・ラグやドラッグ・ロスが生じないように、優れた医薬品を日本で最初に届けられることを目標にさまざまな審査制度が用意されています。
現在の問題はもはや審査期間の長さではなく、ドラッグ・ロス
前述のとおり、日本の審査期間は世界トップクラスのスピードです。近年の問題は開発自体の遅れや日本で開発されないという「ドラッグ・ロス」です。
最近、日本で未承認の薬が増えています。2016年~2020年に欧米で承認された新規薬候補化合物143品目について、日本ではうち86品目が開発未着手であり、その過半数が海外ベンチャー発で、海外のみで開発されています。
その大きな要因は、開発のロードマップに日本が含まれていないことです。海外のベンチャーなど新興企業の薬が海外で第Ⅲ相試験より前に早期承認で進められるなかで、日本が含まれないケースが多い状況です。また、日本の市場規模等の問題で、第Ⅲ相試験に日本が入っていないという現状があります。
このドラッグ・ロスの対策について「創薬力の強化・安定供給の確保等のための薬事規制のあり方に関する検討会」で検討され、2024年4月に報告書が公表されています。その内容には、希少疾病用医薬品の指定のあり方、小児用医薬品の開発促進、日本人第Ⅰ相試験要否の検討、条件付き早期承認制度の在り方などが含まれています。
承認審査では何を決めているか?
非常に大まかにまとめると、承認審査では、「そもそも承認していいかどうか」ということと、「効能・効果と用法・用量(承認事項)等の決定」を行っています。これらと「関連注意」をもって、どんな患者さんに投与できるとするか、どのように投与するか、ということを決めます。
そして、申請資料等を基づき審査を行い、その概略を審査報告書としてまとめます。
承認審査の結果は添付文書に記載されており、効能・効果と用法・用量(承認事項)をはじめ、補足する関連注意や重大な副作用、臨床成績などが示されます。
また、承認後に世に出るその他の文書として、重篤な副作用があり、自覚症状で早期発見が期待できる医薬品を中心に作成される「患者向医薬品ガイド」や、副作用等、薬を使うときに特に注意が必要な内容をわかりやすく説明した「患者向け資材」などがあり、PMDAウェブサイトで確認することができます。
どういう視点で審査しているのか?
審査員のための留意事項は以下のように定められています。こちらは15年以上前に発出されたものですが、現在の審査にも当てはまる内容となっております。
- 実施された試験や提出された資料の信頼性が担保されていること
- 適切にデザイン(計画)された臨床試験結果から、集団における有効性がプラセボ*よりも優れていると考えられること
- 得られた結果に臨床的意義があると判断できること
- ベネフィット(効果)と比較して、許容できないリスク(副作用など)が認められていないこと
- 一定の有効性及び安全性を有する医薬品を恒常的供給可能であること
*プラセボ:有効成分を含まない薬
臨床試験の評価の基本は「比較」
新しい薬の承認は、第Ⅲ相での標準治療との比較が大原則です。
第Ⅱ相試験:第Ⅲ相試験で検証すべき仮説を設定するための情報を得るために計画・実施
第Ⅲ相試験:事前に設定された仮説を検証するために計画・実施
計画や評価は、その試験の位置付けを考えながら検討がなされます。
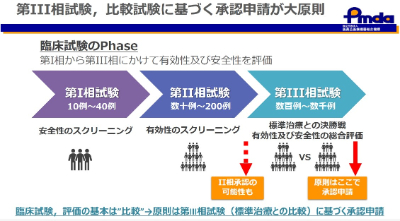
原則は第Ⅲ相試験での比較が必要、ただし、第Ⅱ相試験で承認される場合もある
患者の人数が極端に少ない希少疾患では、比較試験をすることは困難であり、第Ⅱ相試験の結果に基づき承認された薬剤がこれまでも複数あります。ただし、その場合、基礎的な裏付けであったり、第Ⅱ相試験で臨床的に意義がある結果が示されていることは不可欠です。
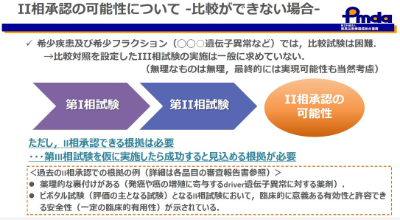
リアルワールドデータ(RWD)とは
比較試験が実施できない場合はどうするのか?その場合の有効性の評価のサポートの位置付けとして、リアルワールドデータ(RWD)の活用が検討されます。RWDとは日常の実臨床の中で得られる医療データの総称です。電子カルテのデータをはじめ、ウェアラブルデバイスから得られるデータなどがあります。
希少疾患などの比較試験を行うことが出来ない場合の評価にRWDの活用が議論されています。ただし、実際には臨床試験の結果と比較可能なデータをRWDで得ることは難しく、承認申請も目的に含めた前向きなデータ収集が望まれます。
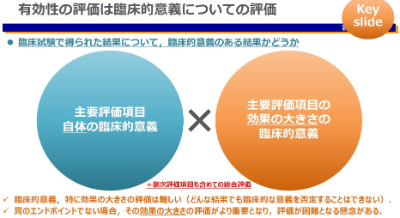
「主要評価項目」とは?
簡単に言えば、それを達成すれば臨床試験成功、と判断できる項目を主要評価項目といいます。臨床的意義があり、その分野で合意を得られていることが望ましいものです。有効性の評価は、得られた臨床試験の結果が臨床的に意義があるかどうかを評価することになります。主に主要評価項目の「項目自体の臨床的意義」と「効果の大きさの臨床的意義」から有効性を評価します。
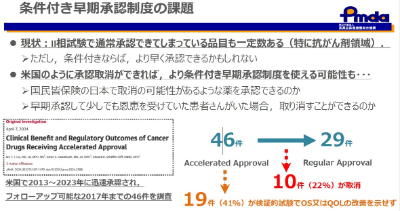
条件付き早期承認制度について
より早く使えるようにするための制度であり、2020年に法制化されています。
この承認には以下の4つの要件があります。
① 疾患の重篤性
② 医療上の有用性
(i)既存治療なし、または(ii)既存治療と比較して優れた有効性、安全性等
③ 第Ⅲ相試験等の比較試験の実施が困難または実施可能でも長期間を要する
④ 比較試験以外の臨床試験等での一定の有効性及び安全性
現時点で、これまで5つの薬がこの制度で承認されており、そのうちのひとつは、デュシェンヌ型筋ジストロフィーの治療薬「ビルテプソ」です。なお、条件付き早期承認制度に該当しなくても第Ⅱ相試験の結果に基づき通常承認することも可能であり、この制度に適した品目がそう多くないこと、現状、早期承認した後の承認取消が日本では馴染みにくいことなどの課題も抱えています。
有効性と安全性の評価はバランスの評価(ベネフィット・リスクバランス)
有効性と安全性の評価は最終的には、ベネフィットとリスクのバランスで評価されます。このバランスは、なかなか数字で明確にできるものでもなく、それぞれの審査品目の疾患の背景なども含めて判断する必要があり、画一的なものではありません。このベネフィット・リスクバランス判断の一助になるかもしれないものとして、「患者にとって何が重要か」、「どれだけ重要か」という要素を科学的に評価する試験について、ICHで現在議論が行われています(ICH E22:「Patient Preference Studies(患者選好試験)に関する全般的考慮事項」)。

講演を終えて:主催者からの提言
手塚先生のご講演は密度があり、これまで多くの患者と家族が知らなかったことをたくさん教えていただきました。
手塚先生は「本当にこの薬でいいのか」を判断するためには「比較が重要」と強調されていました。
わたしたちは日常生活の中で、どんな薬であっても「100パーセント・誰にでも・副作用がなく、効果がある」わけではないと知っています。たとえば頭痛薬を飲むと胃が荒れやすい、ということは、生活の常識としてお持ちでしょう。
「わたしが治った薬だから、あなたも治るんじゃない?」という風説だけでは、本当に正しいかどうかわからないだけでなく、かえって症状を悪化させる可能性があります。
「比較をして確かめる」。PMDAが行う確かなサイエンスは、わたしたちの心と体を守るためのものです。
ただ、それぞれの病気ごとに症状は違います。どんな生活をしているかも違うでしょう。
それなら、「比較」に患者が参加することがもっとも確かではないでしょうか?
患者が「比較」というサイエンスに参加することは、治療薬を早く手に入れるためのひとつの方法です。
そのためには、患者自身がなぜこの病気になるのか、患者として抱えている痛みや苦しみはどんなものかを、世の中の人たちがわかるように説明できることが大事です。
今、世の中で言われている「患者参画」をしている患者たちは、学んでいて、知識を持ち、他人の意見を聴き、そして自らの意見を表明できています。
患者なら誰だって治りたいのは一緒。しかし、そのためにできることに気がつくか・気がつかないかには大きな違いがあります。
PMDAでも、患者参画ガイダンスを用意し、取り組みを進めています。
https://www.pmda.go.jp/files/000242830.pdf
当会はこれからも、患者と家族として、同じ立場のみなさんが患者参画できるよう、さまざまな知識提供を行います。
ぼんやり待つだけではなく、まずは「患者参画」を進めてみませんか。患者であってもできることはたくさんあります。
最後に、多忙な中でご講演をいただきました手塚先生に、厚くお礼を申し上げます。
筋強直性ジストロフィー患者会

